Intellectual Disability (ID) is a debilitating condition with deficits in cognitive functioning (IQ < 70) and adaptive skills. Global developmental delay (DD) is the term used in children < 5 years, who show deficits in two or more developmental domains.
ID is often associated with behavioural problems (such as hyperactivity, autism, aggressive and self-injurious behaviour), epilepsy and other neurological disabilities, resulting in a considerable psychological, social and economic burden to patients and families.
A full definition of ID, by the American Association on Intellectual and Developmental Disabilities (AAIDD), can be found by clicking this link.
A systematic literature review identified 116 inborn errors of metabolism which are causally related to Intellectual Disability and amenable to therapy.
These diseases are presented on this website as an interactive tool for the clinician and scientist, both expert and trainee.
The information is presented in several different ways: ranging from the biochemical categories, signs & symptoms, diagnostic tests, genes, to therapies & evidence.
For each condition a disease page has been created as information portal with access to specific genetics, biochemistry, phenotype, diagnostic tests and therapeutic options. Online Resources include: OMIM, Gene Reviews, Orphanet, Gene Cards, Wikipathways and more.
Treatable diseases should be prioritized in the diagnostic work-up of any individual with with developmental delay or intellectual Disability. Early diagnosis & treatment improves outcomes.

FAQ on Intellectual Disability (ID)?
Intellectual Disability affects 1 - 3% of children and adults worldwide.
Causes of ID are many. Therefore, to identify the origin of ID in a child can pose a huge challenge to doctors. Causes of ID may occur as early as in fetal life or any time later in life. Examples include noxious hits to the fetal brain due to alcohol (fetal alcohol syndrome) or infections during pregnancy or later in life or traumatic head injury. Apart from such environmental and acquired causes, genetic origin is an important cause of ID.
Genetic causes by themselves are manifold. Therefore, a large number of tests are performed and many specialists are involved to elucidate those genetic causes.
Guidelines for assessment of genetic ID used to be based on frequencies of single conditions and on the yield of diagnostic methods and procedures. The wide implementation of exome / genome sequencing had led to identification of new causes of ID. However, about up to 50% of all individuals undergoing genetic testing for ID remain without a diagnosis. Also, despite the high frequency and high diagnostic yield, no causal treatment is available for most of the conditions identified by these investigations.
Inherited metabolic disorders (IMDs) constitute a subgroup of genetic ID, more than 1400 in number and affecting 1 in 700 newborns. IMDs are defined by the international metabolic community in the International Classification of Inherited Metabolic Disorders (ICIMD) as: ‘Any condition in which the impairment of a biochemical pathway is intrinsic to the pathophysiology of the disease, regardless of whether there are abnormalities in currently available biochemical laboratory tests’(www.icimd.org). It is this subgroup, that does include conditions, which are amenable to treatment targeting the pathophysiology.
Despite the option to profoundly improve prognosis by early implementation of therapy, screening for IMDs is not always performed systematically. This is explained by the fact that treatable IMDS are rare and thus not prioritized in the diagnostic work-up. Another reason is the fact that up-to-date protocols summarizing comprehensively all IMDs causing treatable ID and indicating the type and order of tests are not always used.
The number of treatable IDs is increasing. During the past two decades, an increasing number of IMDs have become amenable to treatments, and technologies for better recognition have been introduced into clinical and health care practice.
Therefore, we undertook literature reviews to identify all treatable IMDs with Intellectual Disability as a prominent feature and to characterize types of treatments and evidence for effect. In 2012 the number was 81, increasing to 89 in 2014, and early 2021, we identified as many as such 116 ‘treatable intellectual disabilities’.
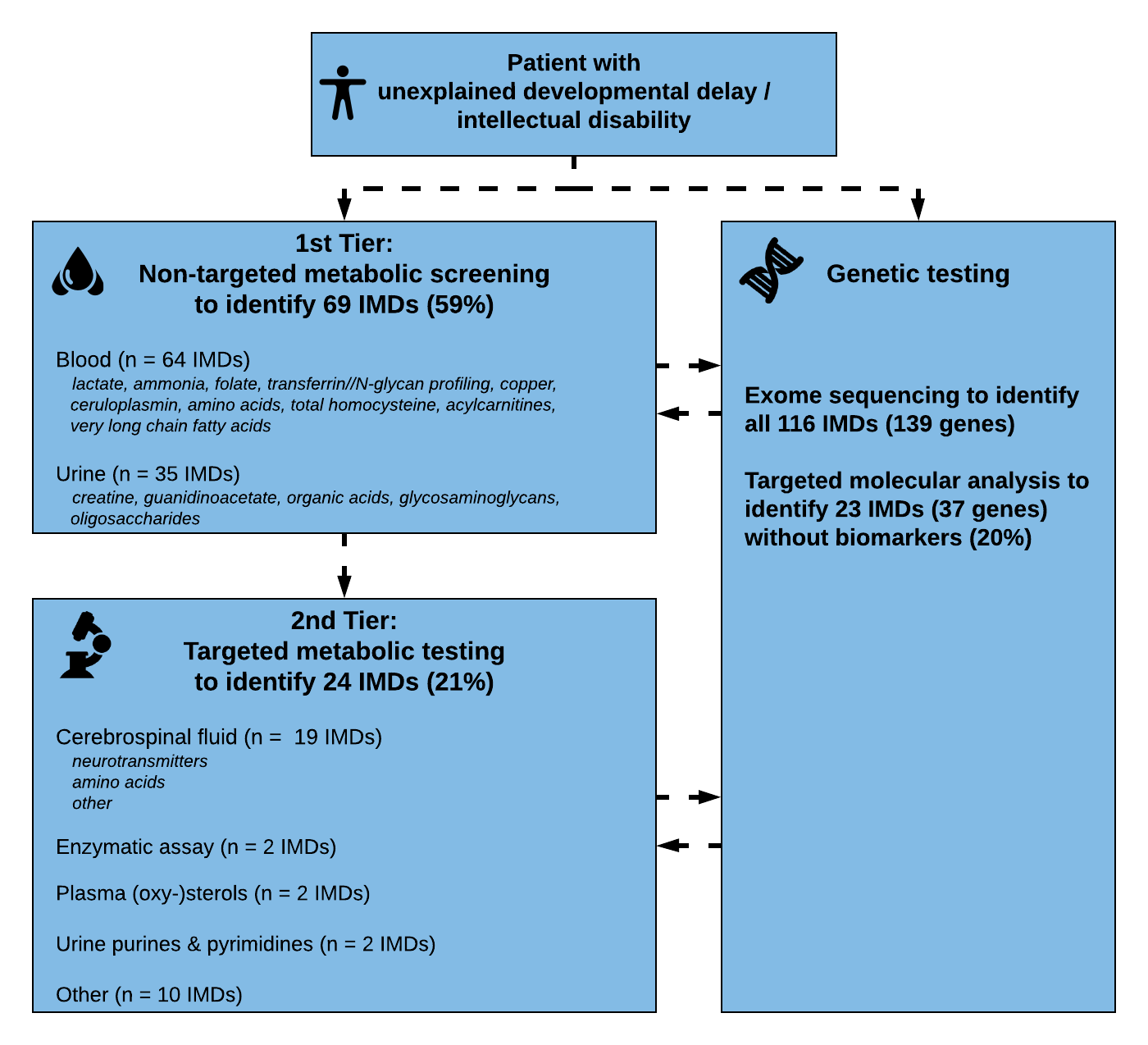
We used our literature review results to update our diagnosed algorithm proposed in 2014. During the past years, exome sequencing has become accepted as first line tier testing in many countries around the world. However, metabolic screening is still applied given the specificity and sensitivity of tests, the short turnaround times and relative affordability and availability.
Metabolic profiles can also serve as functional readout, and “deep metabolic phenotyping” can help in the interpretation of genetic data.
Therefore, we included both strategies in our algorithm to facilitate a practical guide for biochemical and genetic/genomic diagnosis of developmental disability and intellectual disability. We first assessed which tests are necessary to identify each of the conditions. Accordingly, we grouped the IMDs into those diagnosed via ‘metabolic screening tests’ (1st Tier) versus IMDs diagnosed via ‘single test per single disease’ (2nd Tier) approach. First tier screening tests were defined as tests in blood and urine which are readily available in biochemical laboratories in most developed countries. Metabolic tests in the 2nd tier group evaluate Treatable IDs for which biochemical markers are difficult to interpret, and/or conventional diagnostic approach requires an invasive procedure or poorly accessible test (i.e., only performed in few centres worldwide). Furthermore, we analyzed which IMDs have no (reliable) biomarker profile and require primary molecular or (targeted) ES analysis. This approach with different strategies and tiers was then translated into a step-wise algorithm.
We identified treatments varying from nutritional therapy (most frequent), vitamin & trace element enzyme replacement therapy, hematopoietic stem cell transplant, solid organ transplantation, pharmacological therapy, gene-based therapy, other (e.g., hemodialysis). Quality evidence varied from level 1-3 (trials, cohort studies, case-control studies) for 19% and 4-5 (case-report, expert opinion) for 81% of treatments; Therapies with limited evidence can be very effective however, especially if started early in life (creatine substitution in AGAT deficiency for example). Finally, most therapies on our list are accessible, affordable and with acceptable side-effects.
Because Time = Brain
The low frequency of these diseases should not prohibit an active process of identification. If we find more patients with treatable ID, we can improve their outcomes in many ways. In some, particularly young children treatment can halt progression of an otherwise ongoing developmental delay and preserve brain function. Other children may benefit because additional behavioural problems or epilepsy may improve on treatment. Even in the absence of causal therapy, determination of the genetic aetiology of ID is pivotal in generating and translating knowledge relevant to the anticipatory healthcare needs of the individual and their family –including genetic counselling- and minimizing gaps or disparities in care.

The original version of this tool was published in 2012 as part of the TIDE BC program in Vancouver, Canada.